糖化应激和抗衰老
糖化应激与阿尔兹海默症
糖化应激与阿尔兹海默症
痴呆症与阿尔兹海默症
2012年的日本痴呆症老年患者人数估计为462万人(1)。此后仍在继续增长,预计将在2025年达到675万人(患病率18.5%),2050年达到797万人(患病率21.1%)。经计算,到2025年时,65岁及以上的老年人中大约每5人就有1人是痴呆症患者,这是一种任何人都很有可能罹患的疾病(假设各年龄段的痴呆症患病率自2012年以来保持不变)。
痴呆症是指曾经达到正常水平的认知功能,因后天性的脑部疾病而发生持续性的下降,呈现出对日常生活和社交生活造成影响的状态,是一种在没有意识障碍的情况下就能观察到的症状(2)。世界卫生组织(WHO)的国际疾病分类第10版(ICD-10)中,将其定义为“痴呆是由于脑部疾病造成的症候群,通常为慢性或进行性之本质,可造成多种高级皮质功能之障碍,包含记忆、思维、定向感、理解能力、计算、学习能力、语言及判断能力”(2)。痴呆症分为阿尔兹海默型痴呆症(阿尔兹海默症)(Alzheimer’s disease:AD)、脑血管性痴呆症(vascular dementia:VD)、路易体痴呆症(dementia with Lewy bodies:DLB)和其他疾病引起的痴呆症这4种。
在岛根县海士町进行的调查发现,在943名65岁及以上的老年人中,有104人被确认为痴呆症患者,其中AD为63.5%,VD为15.4%,DLB为4.8%,帕金森病痴呆(Parkinson’s disease dementia:PDD)为6.7%,进行性核上性麻痹(progressive supranuclear palsy:PSP)为1.9%,额颞叶变性型痴呆症(frontotemporal lobar degeneration:FTLD)为0.96%,由AD直接引起的痴呆症占到60%及以上(3)。
AD的症状包括“轻度认知功能障碍”(mild cognitive impairment:MCI)和“精神行为症状”(外围症状)(behavioral and psychological symptoms of dementia:BPSD)。MCI的症状有记忆障碍、视空间障碍、判断力下降、失语、失用、失认等,这些症状统称为核心症状(core symptom)。BPSD则包括兴奋、尖叫、情绪不稳、焦躁、游荡、有悖社会文化的不当行为、性欲脱抑制、收集癖、谩骂、尾随、焦虑、抑郁、妄想、幻觉等。BPSD又可进一步分为兴奋、易激惹、焦躁、幻觉、妄想等阳性症状(positive symptoms)和乏力、淡漠、抑郁等阴性症状(negative symptoms)。另外,约有60%~90%的痴呆症患者表现出多种BPSD症状,其中以淡漠、兴奋、易激惹、抑郁等最为常见(4)。
AD的神经病理学特征为在脑内海马体(hippocampus)和大脑皮层(cerebral cortex)中可见萎缩(atrophy),在显微镜下可大范围观察到神经细胞的脱落、老年斑(senile plaques)和神经原纤维缠结(neurofibrillary tangle:NFT)的沉积(图1)(5)。NFT在电子显微镜影像中呈现为一种特殊的纤维束,该纤维束被称为配对螺旋样纤维(paired helical filament:PHF)。老年斑和PHF的主要成分被确认为β-淀粉样蛋白(amyloid-β protein:Aβ)和高度磷酸化的tau蛋白(tau protein)。tau蛋白存在于神经轴突中,是一种分子量约为5万的微管(microtubule)结合蛋白,其具有促进和稳定微管聚合的作用。微管构成细胞骨架,在细胞内的蛋白和细胞内细胞器中发挥着运输通道的作用。这一细胞内运输机制会在tau蛋白磷酸化后,因微管失稳而遭到抑制。
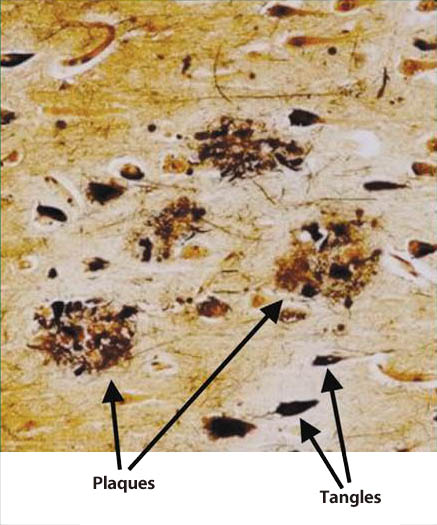
图1. 阿尔兹海默症患者脑内的老年斑和神经原纤维缠结(5)
plaques:老年斑, tangles:神经原纤维缠结
另外,Aβ沉积形成的老年斑,对于AD的疾病特异性要高于NFT。弥漫性老年斑(diffuse plaques)是AD大脑的最早期病变,其主要由非纤维性Aβ沉积形成。在表现为常染色体显性遗传的家族性AD中发现,Aβ前体(amyloid precursor protein:APP)的点突变(point mutation)和重复(duplication)与该疾病相关。在Aβ中,聚合的Aβ寡聚体(amyloid-β aggregate)尤其具有神经毒性。由此可推断出,在AD的发病机理中,Aβ的脑内沉积要早于tau蛋白,是一种与AD病因密切相关的现象。围绕Aβ展开的AD发病机制学说,被称为“淀粉样蛋白级联假说(amyloid cascade hypothesis)”(6)。
另外,与正常对照组相比,AD患者大脑皮层中的乙酰胆碱转移酶(choline acetyltransferase:ChAT)活性下降。AD病患者大脑皮层的ChAT活性与总体认知评估分数(global cognitive score)相关。在AD的基底前脑(basal forebrain)的Meynert基底核(nucleus basalis of meynert:NBM)中,能够观察到胆碱能神经元的显著性脱落,从位于基底前脑的中枢神经系统细胞群投射(projection)至大脑皮层和海马体的胆碱能神经通路出现紊乱。AChE抑制剂和尼古丁(nicotine)能够促进脑内乙酰胆碱系统的神经传导。但是,研究证实,如果使用阿托品(atropine)和东莨菪碱(scopolamine)来阻断脑内乙酰胆碱系统的神经传导,会对学习和记忆行为造成影响。由此可见,胆碱能神经障碍与包括学习记忆在内的认知功能密切相关。这种因胆碱生成不足而引发AD的发病机制学说,被称为“胆碱能假说(cholinergic hypothesis)”(7)。
此外,基于AD患者的老年斑沉积与神经细胞脱落并不相关这一观察结果,还有一种学说认为,痴呆症的主要致病物质是tau蛋白,作为微管稳定因子的tau蛋白消失后,神经细胞的轴突处便会形成配对螺旋样纤维(paired helical filament:PHF),产生NFT,从而导致细胞骨架的变性。这种AD发病机制学说被称为“tau假说(tau hypothesis)”(8-9)。不过,对于tau蛋白的过度磷酸化和PHF的形成,哪个先发生,目前则仍存在争议。
从蛋白质变性、交联反应增强和RAGE介导诱发炎症的观点来看,糖化主要与淀粉样蛋白级联假说和tau假说有关。
阿尔兹海默症与糖尿病
糖尿病的典型并发症有神经疾病(neuropathy)、视网膜病变(retinopathy)、肾病(nephropathy)、心血管疾病(cardiovascular disease)等。此外,在糖尿病患者中痴呆症发病率较高,这一点也广为人知(图2)(10)。鹿特丹大学在自1990年起的两年间,开展了一项大规模流行病学研究(Rotterdam study),针对平均年龄为55岁及以上的6370名患者(2型糖尿病患者(2DM)692名、非糖尿病患者5678名),调查了糖尿病与痴呆症的相关性(11)。结果显示,有126人(2.0%)发病痴呆症,其中27.0%为2DM。这126名痴呆症发病者中,有89人(70.6%)为AD;未发病痴呆症的6244人中10.5%为2DM。由此结果推算出了,2DM与非糖尿病患者相比,AD发病风险是其1.9倍。
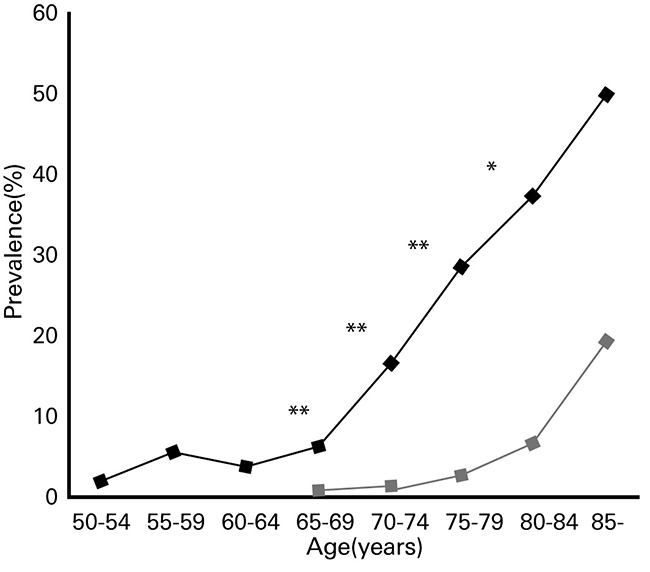 图2. 糖尿病患者和健康人在不同年龄段的痴呆症患病率
图2. 糖尿病患者和健康人在不同年龄段的痴呆症患病率
◆:糖尿病患者, ■:健康人
对于糖代谢能力与痴呆症的关系,曾在福冈县久山町进行过一项长期性的调查(12)。在1988年至2003年的15年间,对1017名未患痴呆症的年龄为60岁及以上的老年人(女性580人、男性487人)进行追踪调查,在调查开始时,对232人(女性153人、男性79人)进行了75 g口服葡萄糖耐量试验(oral glucose tolerance test:OGTT),用由此得到的糖代谢能力评价结果,研究了该评价结果与痴呆症发病之间的关系。结果显示,关于痴呆症发病风险(调整性别和年龄后),若将糖耐量正常组(control)作为1.0(n=115),则空腹血糖受损组(impaired fasting glycemia)为0.63(n=13), 糖耐量减低组(impaired glucose tolerance)为1.35(n=63),糖尿病组(diabetes)为1.74(n=41),亦即糖代谢能力越低,发病风险越高。由此推测,糖尿病和糖代谢能力与痴呆症发病相关。
阿尔兹海默症与AGEs
AD患者大脑的神经病理学特征是脑萎缩、老年斑和神经原纤维缠结。老年斑由β-淀粉样蛋白(amyloid-β protein:Aβ)构成,是一种大约由40个氨基酸组成的肽(Aβ40、Aβ42),分子量为4300~4500。Aβ是在β-分泌酶和γ-分泌酶(secretase)作用下,从淀粉样前体蛋白(amyloid-β protein precursor:APP)中分解出来的。在AD患者体内,Aβ会凝集,并形成不溶性纤维,以淀粉样沉积。在葡萄糖的环境中培养后,Aβ发生糖化和AGE化(AGE-Aβ)。Aβ的氨基酸序列中,第16位和第28位有赖氨酸残基(Lys:K),第5位有精氨酸残基(Arg:R),这些氨基酸序列位点可能与糖化有关。此外,在葡萄糖的环境中培养,Aβ会发生凝集。与未添加AGE-Aβ时相比,向Aβ-葡萄糖溶液中添加AGE-Aβ后,凝集被大大促进(13)。由此推测出,AGE-Aβ可以起到促进Aβ凝集的“种子(seed)”作用。此外,在额叶中含有老年斑的组织的蛋白中的AGEs量,AD患者要比健康老年人高出3倍(14)。但是,在Aβ-葡萄糖反应系统中添加糖化反应抑制剂氨基胍(aminoganidine)进行培养,会对Aβ的凝聚形成产生抑制(15)。另外,现已证实老人斑中还存在戊糖素、吡咯素和CML(14-15)。由此推测,因糖化而引起的Aβ的AGE化,与蛋白交联的形成有关,是Aβ凝集的促进因素之一。
从AD患者大脑中取得的配对螺旋样纤维(paired helical filament:PHF)的tau蛋白已发生AGE化。但是从非AD患者或没有患痴呆症的大脑中取得的可溶性tau蛋白并未观察到AGE化的迹象。在tau蛋白的in vitro实验系统中,发生糖化的tau蛋白形成PHF样纤维。而没有发生糖化的tau蛋白不会形成纤维(16)。PHF中含有戊糖素、吡咯素和CML。并且,tau蛋白的氨基酸序列中有13个赖氨酸残基,其中有6个发生了糖化(17)。tau蛋白的糖基化位点正是它与微管的结合位点,因而导致结合功能受损。糖化tau蛋白诱发活性氧、IL-6等的氧化应激,使神经细胞功能受损(18)。糖化tau蛋白诱发淀粉样前体蛋白(amyloid-β protein precursor:APP)和Aβ形成。tau蛋白因磷酸化和糖化而发生微管结合功能下降,作为tau蛋白原有功能的细胞内传导能力降低。因此,细胞无法将APP分泌到细胞外,细胞内APP蓄积。通过上述机制,Aβ和tau蛋白的糖化促进了Aβ的沉积和凝集,同时也增强了tau蛋白的沉积。
此外,AD患者大脑内的小胶质细胞(microglia)和星形胶质细胞(astrocyte)中所含的戊糖素、CML以及作为AGE受体的RAGE(receptor for AGEs)(19),与健康老年人相比显著性增加。小胶质细胞是一种存在于中枢神经系统的细胞,又称常驻巨噬细胞(macrophage),具有清除和修复神经元等的功能(20)。另一方面,已证实小胶质细胞的激活可导致肿瘤坏死因子(TNF-α)、白介素(IL)-1β、干扰素(IFN)等炎性细胞因子的形成,表明它可能对神经细胞存在损害作用(21)。星形胶质细胞是一种只在脑和脊髓中存在的星形神经胶质细胞,在维持中枢神经系统功能方面具有调节血流、为神经细胞供给能量、干预突触功能、调节神经递质等作用(22)。向小胶质的培养细胞中添加AGE化鸡卵清蛋白(chicken egg albumin-AGE)后,可观察到RAGE介导的NO(nitric oxide)、IL-6、TNF-α等物质的产生,并会诱发炎症(23)。另外,在向大鼠大脑皮层神经元细胞(cortical neuronal cell)培养系统添加各种AGEs的实验中,证实了与Amadori 化合物(amadori products)、乙醇醛(glycolaldehyde)衍生的AGEs、甲基乙二醛(methylglyoxal)衍生的AGEs、乙二醛(glyoxal)衍生的AGEs相比,甘油醛(glyceraldehyde)衍生AGEs具有更强的诱发凋亡(apotosis)作用(24)。此时,CML和CEL均未诱发细胞死亡。由此推测出,随着Aβ和老年斑在大脑内的不断蓄积而发生的AGEs生成和蓄积,能够诱导Aβ出现神经毒性,诱发RAGE介导的炎症和凋亡,并参与AD病情的神经细胞死亡的促进和增强(图3)(25)。
上述内容表明,灵活使用可减少糖化所导致的AGEs形成及蓄积的物质,或会成为防止AD发病的有效措施(26-27)。
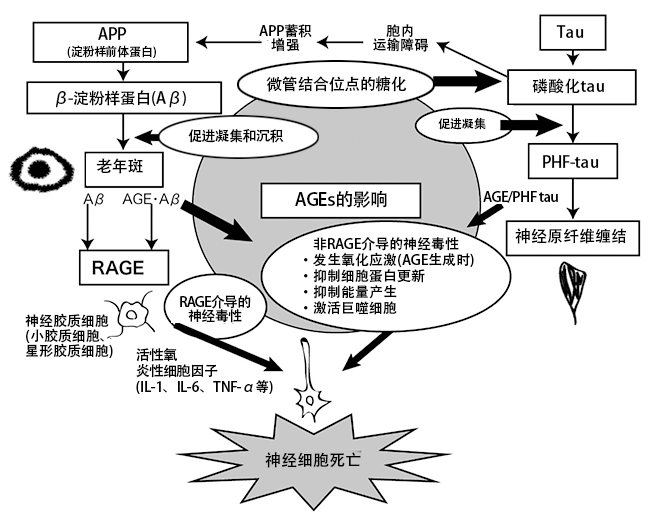
图3. AGEs对阿尔兹海默症的影响(25)
References
-
- 二宮利治:日本における認知症の高齢者人口の将来推計に関する研究総括研究報告書, 厚生労働省; 2015.
- 認知症疾患治療ガイドライン2010 ; 日本神経学会監修.
- Wada-Isoe K, et al.:Neuroepidemiology. 2009; 32: 101–106.
- Ileda M et al.:J Neurol Neurosurg Psychiatry. 2004; 75: 146–148.
- Blennow K, et al.:Lancet. 2006; 368: 387–403.
- Karran E, et al.:Nature Reviews Drug Discovery. 2011; 10: 698-712.
- Francis PT, et al.:J Neurol Neurosurg Psychiatry. 1999; 66: 137–147.
- Mudher A, et al.:Trends Neurosci. 2002 ; 25(1): 22-26.
- Maccioni RB, et al.:Archives of Medical Research. 41; 2010: 226-231.
- Kimura R, et al.:Psychogeriatrics. 2008; 8: 73-78.
- Ott A, et al.:Neurology 53(9); 1999: 1937-1942.
- Ohara T, et al.:Neurology. 2011; 77: 1126–1134.
- Vitek MP, et al.:Proc Natl Acad Sci USA. 1994; 91: 1994.
- Smith MA, et al.:Proc Natl Acad Sci USA. 1994; 91: 5710-5714.
- Horie K, et al.:Biochem Biophys Res Commun, 1997; 236: 327–332. 1997
- Ledesma MD, et al.:J Biol Chem. 1994; 269: 21614-21619.
- Nacharaju P, et al.:J Neurochem. 1997; 69: 1709-1719.
- Yan SD, et al.:Proc. Nati. Acad. Sci. USA: 1994: 7787-7791
- Srikanth V, et al.:Neurobiology of Aging. 2011; 32: 763–777.
- Takeda A, et al.:Acta Neuropathol. 1998; 95: 555–558.
- 錫村明生:福岡医誌. 100; 243-247 2009
- Hamby ME, et al.:Neurotherapeutics. 2010; 7: 494-506.
- Dukic-Stefanovic S, et al.:Journal of Neurochemistry. 2003; 87: 44–55.
- Takeuchi M, et al.:J. Neuropathol. Exp. Neurol. 2000; 59: 1094–1105.
- 佐々木信幸:AGEsとアルツハイマー病, AGEsの研究の最前線. 2004: 171-177.
- Dukic-Stefanovic C, et al.:Biogerontology. 2001; 2: 19–34.
- Munch G, et al.:Biochem Soc Trans. 2003; Pt 6: 1397-1399.
糖化应激和抗衰老
- 什么是糖化应激?
- 糖化应激标志物的检测方法(1) 血糖、糖化蛋白、糖化反应中间体的检测
- 糖化应激标志物的检测方法(2) AGEs的检测
- 糖化应激标志物的检测方法(3) 抗糖化作用评价
- 糖化应激和AGEs受体
- 糖化应激与肾疾病
- 糖化应激和皮肤老化
- 糖化应激与动脉硬化
- 糖化应激和精神分裂症
- 糖化应激与肝病
- 糖化应激与不孕症
- 糖化应激与阿尔兹海默症
- 糖化应激对策(1) 血糖控制
- 糖化应激对策(2) 糖化反应的抑制
- 糖化应激对策(3) AGEs的降解和排泄
- 糖化应激对策(4) 膳食来源的AGEs
- 糖化应激对策的课题和展望