Glycative stress and Anti-aging
Glycative stress and Alzheimer's disease
Glycative stress and Alzheimer’s disease
Dementia and Alzheimer’s disease
In Japan, the number of elderly people with dementia was estimated to be 4.62 million in 2012 1). The number has been increasing since then and it is projected to reach 6.75 million (prevalence of 18.5%) in 2025 and 7.97 million (prevalence of 21.1%) in 2050. This means that by 2025, about 1 in 5 elderly people over the age of 65 will have dementia, which is a disease that can affect anyone. (This is based on the assumption that the prevalence of dementia in each age group will remain constant after 2012.)
Dementia indicates a condition in which the persistence of the cognitive function that had once reached normal levels has declined due to acquired brain damage and thus disrupts a person’s daily life and social life, and refers to the symptoms that are observed when impaired consciousness is not present 2). According to the 10th edition of the International Classification of Diseases (ICD-10) by the World Health Organization (WHO), it is “a syndrome due to disease of the brain, usually of a chronic or progressive nature, in which there is disturbance of multiple higher cortical functions, including memory, thinking, orientation, comprehension, calculation, learning capacity, language and judgement.” 2). Dementia is classified into 4 types: Alzheimer’s disease (AD), vascular dementia (VD), dementia with Lewy bodies (DLB), and dementia due to other diseases.
A survey conducted in Ama Town, Shimane Prefecture found that dementia was observed in 104 out of 943 people aged 65 and older. Among all the types of causes, dementia directly caused by AD accounted for more than 60% of the cases with AD 63.5%, VD 15.4%, DLB 4.8%, Parkinson’s disease dementia (PDD) 6.7%, progressive supranuclear palsy (PSP) 1.9% and frontotemporal lobar degeneration (FTLD) 0.96% 3).
Symptoms of AD include “mild cognitive impairment (MCI)” and “behavioral and psychological symptoms of dementia (BPSD)”. The symptoms of MCI are memory disorder, disorientation, impaired judgment, aphasia, apraxia, and agnosia, which are collectively referred to as the core symptoms. BPSD includes excitement, screaming, restlessness, impatience, wandering, socio-culturally inappropriate behavior, sexual disinhibition, collectomania, abusive speech, stalking, anxiety, depression, delusions, and hallucinations. Furthermore, BPSD is divided into positive symptoms such as excitement, irritability, impatience, hallucinations and delusions, and negative symptoms such as apathy, indifference and depression. Approximately 60 to 90% of dementia patients have multiple BPSD symptoms, especially indifference, excitement, irritability, and depression 4).
As a neuropathological feature of AD, atrophy is observed in the hippocampus and cerebral cortex in the brain, and neuronal loss and the deposition of senile plaques and neurofibrillary tangle (NFT) are widely observed microscopically (Fig. 1) 5). NFT, which is found under an electron microscope, is a unique fiber bundle called paired helical filament (PHF). Amyloid-β protein (Aβ) and hyperphosphorylated tau protein have been identified as primary structural components of senile plaques and PHF. Tau protein is a microtubule-associated protein with a molecular weight of about 50,000 that exists in the nerve axons, and has the functions of promoting and stabilizing the polymerization of microtubules. Microtubules form a cytoskeleton and function as a transport rail for intracellular proteins and intracellular organelles. When tau protein is phosphorylated, microtubules are destabilized, causing the suppression of the intracellular transport mechanism. 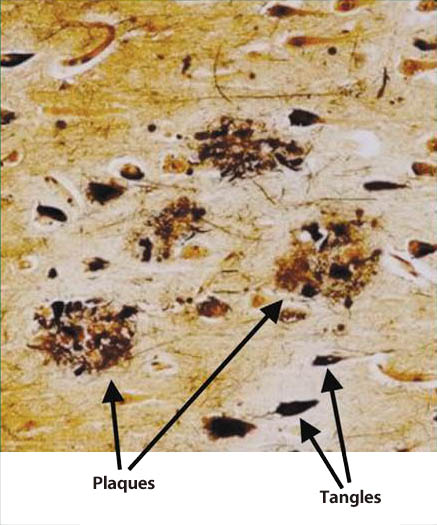
Fig. 1 Senile plaques and neurofibrillary tangles in the brain
of an Alzheimer’s disease patient(5)
Plaques: senile plaques Tangles: neurofibrillary tangles
Also, senile plaques, which are depositions of Aβ, have higher disease specificity to AD than NFT. Diffuse plaques, which are primarily non-fibrillar Aβ depositions, are lesions at the earliest stage of the AD brain.In the familial AD of the autosomal dominant inheritance forms, point mutations and duplications of amyloid precursor protein (APP) have been identified as being associated with the disease. Among Aβ, polymerized amyloid-β aggregates, in particular, have neurotoxicity. These characteristics suggest that deposition of Aβ in the brain precedes tau protein in the onset of AD and is closely related to the pathogenesis of AD. The theory of pathogenetic mechanism of AD which focuses on Aβ is called the “amyloid cascade hypothesis” 6).
Also, AD patients have reduced choline acetyltransferase (ChAT) activity in the cerebral cortex compared to the normal control group. The cerebral cortical ChAT activity in patients with AD correlates with with global cognitive score. In AD, a significant loss of cholinergic neurons are observed in the nucleus basalis of meynert (NBM) of the basal forebrain, and there is a disorder in the cholinergic pathways that project from the central nervous system cells present in the basal forebrain to the cerebral cortex and hippocampus. AChE inhibitors and nicotine boost acetylcholine neurotransmission in the brain. On the other hand, it has been demonstrated that blocking acetylcholine neurotransmission in the brain by administration of atropine and scopolamine affects learning and memory behavior. These facts suggest that cholinergic (acetylcholine) nerve dysfunction is closely associated with cognitive functions including learning and memory. The theory on the pathogenesis of AD that is due to the failure in production of acetylcholine is called the “cholinergic hypothesis” 7).
In addition, based on the observation that senile plaque deposition in AD patients is not correlated with neuronal loss, there is another theory that considers that tau protein is the main causative agent of dementia. The disappearance of tau protein, a microtubule-stabilizing factor, and the formation of paired helical filament (PHF) in the axons of neurons generates NFT, leading to denaturation of the cytoskeleton, which is presumed to be the pathogenosis of dementia.This theory on the onset mechanism of AD is called the “tau hypothesis” 8-9). However, there is some debate as to whether the hyperphosphorylation of tau protein occurs first or the formation of PHF occurs first.
Glycation is mainly related to the amyloid cascade hypothesis and the tau hypothesis from the viewpoint of protein denaturation, enhanced crosslinking and RAGE-mediated induction of inflammation.
Alzheimer’s disease and diabetes
Typical complications of diabetes include neuropathy, retinopathy, nephropathy, and cardiovascular disease. Furthermore, the incidence rate of dementia is known to be high in diabetic patients (Fig. 2) 10).A large-scale epidemiological study conducted by the University of Rotterdam (Rotterdam study) for approximately 2 years from 1990 examined the relationship between diabetes and dementia among 6370 patients (692 patients with type 2 diabetes (2DM) and 5678 non-diabetic patients) with an average age of 55 years and older 11). It found that 126 people (2.0%) developed dementia, out of which 27.0% had 2DM. Among the 126 people who developed dementia, 89 (70.6%) had AD, and of the 6244 people who did not develop dementia, 10.5% had 2DM. Based on this result, the risk for 2DM patients was estimated to be 1.9 times higher in AD than non-diabetic patients.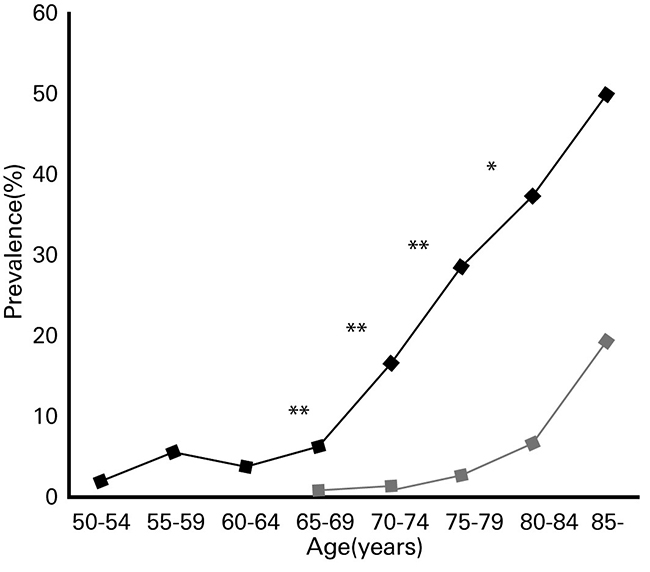
Fig. 2. Prevalence of dementia among diabetic patients and healthy subjects by age group
◆: Diabetic patients ■: Healthy subjects
A long-term study was conducted to examine the relationship between glucose metabolic capacity and dementia in Hisayama Town, Fukuoka Prefecture 12). The study followed 1,017 elderly people aged 60 and over (580 females, 487 males) without dementia for 15 years from 1988 to 2003 and examined the relationship between the results of glucose metabolic capacity evaluations and the onset of dementia by conducting the 75 gram oral glucose tolerance test (OGTT) on 232 (153 females, 79 males) at the start of the study. The results showed that the risk of dementia (after adjustment of gender and age) was 0.63 for the impaired fasting glycemia group (n=13), 1.35 for the impaired glucose tolerance group (n=63) and 1.74 for diabetes group (n=41), when the normal glucose tolerance group (control) was set to 1.0 (n=115), indicating that the lower the glucose metabolic capacity was, the higher risk was. This suggests that diabetes and glucose metabolic capacity are involved in the development of dementia.
Alzheimer’s disease and AGEs
An AD patient’s brain is neuropathologically characterized by brain atrophy, senile plaques, and neurofibrillary tangles. Senile plaques are composed of amyloid-β protein (Aβ), which is a peptide (Aβ40, Aβ42) composed of around 40 amino acids that have a molecular weight of 4,300 to 4,500.Aβ is derived from the amyloid-β protein precursor (APP) by the actions of β- and γ-secretase. In AD brains, Aβ forms aggregates into insoluble fibers and deposits as amyloid.When incubated in the presence of glucose, Aβ is glycated and converts into AGE (AGE-Aβ). The amino acid sequence of Aβ includes lysine residues (Lys: K) at positions 16 and 28 and arginine residues (Arg: R) at position 5 and these amino acid sequence sites may be involved in glycation. In addition, Aβ aggregates when incubated in the presence of glucose.When AGE-Aβ was added to an Aβ-glucose solution, aggregation was accelerated compared to a case where AGE-Aβ was not added 13). The above suggests that AGE-Aβ acts as a “seed” that promotes the aggregation of Aβ. In addition, the amount of AGEs in the tissue protein fraction containing senile plaques in the frontal lobe was three times higher in AD patients than in healthy elderly subjects 14). On the other hand, adding aminoguanidine, a glycation reaction inhibitor, to the Aβ-glucose reaction system and incubating it led to the formation of Aβ aggregates being suppressed 15). Also, the presence of pentosidine, pyrraline, and CML has been observed in senile plaques 14-15). The above suggests that the conversion of AGEs from Aβ via glycation may be related, as one of the factors that promotes the formation of protein crosslinks and the aggregation of Aβ.
Tau protein in the paired helical filament (PHF) obtained from an AD patient’s brain is converted into AGEs. On the other hand, the soluble tau protein obtained from the brain of a non-AD patient or the brain of a non-dementia patient is not converted into AGEs. In an in vitro experimental system of tau protein, glycated tau protein forms PHF-like fibers, while non-glycated tau protein does not form fibers 16). PHF contains pentosidine, pyrraline and CML. Additionally, the amino acid sequence of tau protein contains 13 lysine residues, 6 of which are glycated 17). Since the glycation site of tau protein corresponds to the binding site (binding with microtubules), the binding function is impaired. Glycated tau protein induces oxidative stress such as active oxygen and IL-6, which impairs the neuronal cell functions 18). Glycated tau protein induces the formation of amyloid-β protein precursor (APP) and Aβ. Phosphorylation and glycation of tau protein lowers the microtubule binding function and that leads to a reduction in the intracellular transport function, which is the original function of tau protein. This disables the cells ability to secrete APP externally, resulting in an accumulation of APP inside the cells. Through these mechanisms, glycation of Aβ and tau protein promotes the deposition and aggregation of Aβ, and at the same time accelerates the deposition of tau protein.
Furthermore, pentosidine, CML, and RAGE, which is a receptor for AGEs are found in microglia and astrocytes in the brain of AD patients 19), which have increased significantly compared to healthy elderly people. Microglia, also referred to as resident macrophage, are a type of cell that exist in the central nervous system, and have functions such as removing/repairing neurons 20). On the other hand, activation of microglia has indicated production of proinflammatory cytokines such as tumor necrosis factor (TNFα), interleukin (IL)-1β, and interferon (IFN), suggesting that it may cause damage to neurons 21). Astrocytes are star-shaped glial cells that are specifically present in the brain and spinal cord. They maintain the functions of the central nervous system such as regulation of blood flow, supply of energy to neurons, involvement in synaptic functions and regulation of neurotransmitters 22). When chicken egg albumin-AGE was added to cultured microglial cells, production of nitric oxide, IL-6, TNF-α, etc. via RAGE was observed, causing inflammation 23). Also, in experiments where various AGEs were added to the culture systems of the cortical neuronal cells of rats, a stronger induction of apotosis was observed in glyceraldehyde-derived AGEs compared to amadori products, glycolaldehyde-derived AGEs, methylglyoxal-derived AGEs and glyoxal-derived AGEs 24).On the other hand, CML and CEL did not induce cell death. Based on these results, it is presumed that the formation and accumulation of AGEs associated with the accumulation of Aβ and senile plaques in the brain induces Aβ neurotoxicity, RAGE-mediated inflammation and apotosis, and is involved in the promotion and increase of neuronal cell death, which is a pathological condition of AD. (Fig. 3) 25).
These findings suggest that the use of substances that reduce the formation and accumulation of AGEs due to glycation may serve as measures to prevent the onset of AD 26-27).
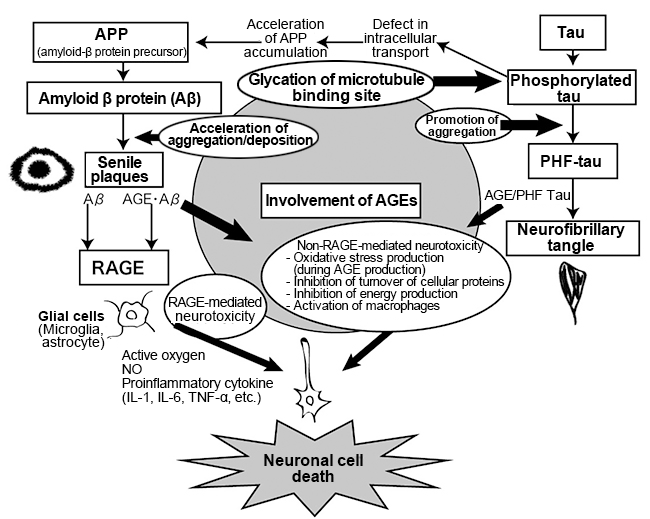
Fig. 3. Involvement of AGEs in Alzheimer’s disease 25)
References
-
- 二宮利治:日本における認知症の高齢者人口の将来推計に関する研究総括研究報告書, 厚生労働省; 2015.
- 認知症疾患治療ガイドライン2010 ; 日本神経学会監修.
- Wada-Isoe K, et al.:Neuroepidemiology. 2009; 32: 101–106.
- Ileda M et al.:J Neurol Neurosurg Psychiatry. 2004; 75: 146–148.
- Blennow K, et al.:Lancet. 2006; 368: 387–403.
- Karran E, et al.:Nature Reviews Drug Discovery. 2011; 10: 698-712.
- Francis PT, et al.:J Neurol Neurosurg Psychiatry. 1999; 66: 137–147.
- Mudher A, et al.:Trends Neurosci. 2002 ; 25(1): 22-26.
- Maccioni RB, et al.:Archives of Medical Research. 41; 2010: 226-231.
- Kimura R, et al.:Psychogeriatrics. 2008; 8: 73-78.
- Ott A, et al.:Neurology 53(9); 1999: 1937-1942.
- Ohara T, et al.:Neurology. 2011; 77: 1126–1134.
- Vitek MP, et al.:Proc Natl Acad Sci USA. 1994; 91: 1994.
- Smith MA, et al.:Proc Natl Acad Sci USA. 1994; 91: 5710-5714.
- Horie K, et al.:Biochem Biophys Res Commun, 1997; 236: 327–332. 1997
- Ledesma MD, et al.:J Biol Chem. 1994; 269: 21614-21619.
- Nacharaju P, et al.:J Neurochem. 1997; 69: 1709-1719.
- Yan SD, et al.:Proc. Nati. Acad. Sci. USA: 1994: 7787-7791
- Srikanth V, et al.:Neurobiology of Aging. 2011; 32: 763–777.
- Takeda A, et al.:Acta Neuropathol. 1998; 95: 555–558.
- 錫村明生:福岡医誌. 100; 243-247 2009
- Hamby ME, et al.:Neurotherapeutics. 2010; 7: 494-506.
- Dukic-Stefanovic S, et al.:Journal of Neurochemistry. 2003; 87: 44–55.
- Takeuchi M, et al.:J. Neuropathol. Exp. Neurol. 2000; 59: 1094–1105.
- 佐々木信幸:AGEsとアルツハイマー病, AGEsの研究の最前線. 2004: 171-177.
- Dukic-Stefanovic C, et al.:Biogerontology. 2001; 2: 19–34.
- Munch G, et al.:Biochem Soc Trans. 2003; Pt 6: 1397-1399.
Glycative stress and Anti-aging
- What is glycative stress?
- Glycative stress biomarker measurement method (1) Measurement of blood glucose, glycated protein and glycation reaction intermediate
- Glycative stress biomarker measurement method (2)AGEs measurement
- Glycative stress biomarker measurement method (3) Evaluation of anti-glycative effects
- Glycative Stress and AGEs Receptors
- What is kidney disease?
- Glycative Stress and Skin Aging
- Glycative stress and arteriosclerotic disease
- Glycative stress and schizophrenia
- Glycative stress and liver disease
- Glycative stress and infertility
- Glycative stress and Alzheimer’s disease
- Glycative stress countermeasures (1) Blood glucose control
- Glycative stress countermeasures (2) Inhibition of glycation reaction
- Measures against glycative stress (3) Degradation and excretion of AGEs
- Measures against glycative stress (4) AGEs contained in food
- Issues and prospects of glycative stress countermeasures
- Issues and prospects of glycative stress countermeasures
- Issues and prospects of glycative stress countermeasures