学術情報
糖化と糖尿病合併症
糖化と糖尿病合併症
糖尿病とは
糖尿病は血液中のグルコース(血糖)が高い状態になって持続し、口渇、多尿、体重減少などをきたす代謝性疾患である。
我々は食物中の栄養素である炭水化物、脂質、たんぱく質を消化・吸収することで、生命を維持し活動するためのエネルギーを得ており、そのうち最もよく使われるのが炭水化物である。炭水化物は消化・吸収された後、グルコースとなって肝臓へ送られ、その一部が脳や筋肉で利用され、残りが肝臓内にグリコーゲンとして蓄えられる。さらに余った分は脂肪になる。
身体活動で血液中のグルコースを消費すると、グリコーゲンが分解されて再びグルコースになって血液中に放出される。このようにして、活動のためのエネルギーが常に維持され、血糖値は一定範囲内の変動におさまっている。
また、細胞が血液中からグルコースを取り込んでエネルギーとして利用するのを助けているホルモンが、膵臓から分泌されるインスリンである。膵臓のインスリン分泌能力の低下や細胞のインスリンに対する感受性が悪くなると、インスリンの作用が低下してグルコースを利用できなくなり、血糖値が高くなる。これを「高血糖」といい、糖尿病ではこの状態が継続する。
糖尿病は血糖検査で確認され、(1)随時血糖値が200mg/dL以上、(2)空腹時血糖値が126mg/dL以上、(3)75gブドウ糖負荷試験で2時間値が200mg/dL以上のいずれかに該当すれば「糖尿病型」と判定される(図1)。さらにHbA1cが6.5%以上、のどの渇きや多飲・多尿、体重減少など糖尿病に特徴的な症状、合併症があれば「糖尿病」と診断される。
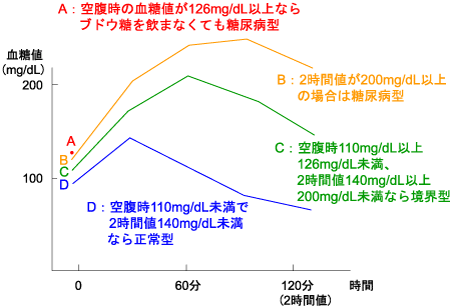
図1. 75gブドウ糖負荷試験による糖尿病型の判定
高血糖は特に神経と血管を中心とした臓器に大きな障害をもたらし、神経障害、網膜症、腎症などさまざまな合併症を発症させる。
合併症の発症・進展要因の一つに、生体タンパク質の糖化進行とAGEsの蓄積がある。Kusunokiらは糖尿病患者110名の血中3DG(3-deoxyglucosone)濃度と合併症進展状態の調査から、100nmol/Lの増加は、腎症リスクを1.94倍、網膜症リスクを2.04倍増加させると報告している。
糖尿病の治療は血糖コントロールにより高血糖を是正し、高血糖によって起こるさまざまな合併症の発症・進行を予防することがすべての基本になっている。
糖尿病性神経障害
糖尿病性神経障害は糖尿病の発症後、比較的早期に出現する合併症で、糖尿病患者の約30%が罹患していると考えられている。神経障害は末梢神経、脳神経、自律神経を侵し、全身症状や機能異常をもたらしている。
高血糖は神経組織のミエリン蛋白、チュブリン、ニューロフィラメントや、細胞外基質タンパク質であるコラーゲン、ラミニン、フィブロネクチンなどを糖化し、3DG、MG(methylglyoxal)、CML(carboxymethyllysine)、CEL(carboxyethyllysine)などのAGEを生成する。このためタンパク質のAGE化は肥厚や多層化を起こす一因になり、軸索内の物質輸送異常や荷電状態の変化、血管透過性の亢進によるバリアーの破綻へとつながっている。
抗CML抗体による免疫染色像の観察から、ヒト末梢神経組織では神経周膜細胞、血管基底膜、軸索、Schwann細胞でAGEsの局在が確認されている。同様に抗RAGE抗体による検討から、神経周囲膜や神経内の血管内皮細胞でAGEs受容体(RAGE)の局在が確認されている。
AGEsが神経障害を発症する機序は、タンパク質のAGE化そのものが神経機能を障害するメカニズムと、AGEs受容体を介して組織反応を惹起する経路が考えられている(図2)。
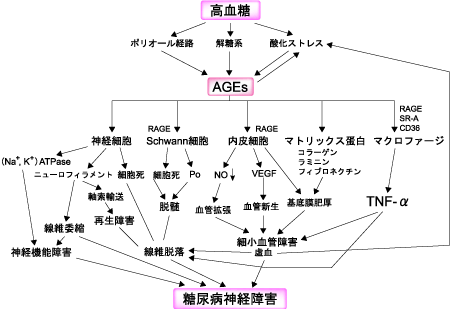
図2. AGEsによる神経障害のメカニズム
神経細胞やSchwann細胞に対する影響としては、AGEsが細胞死(アポトーシス)を引き起こすことが知られており、細胞死が神経線維脱落や脱髄に関連すると考えられている。
RAGEを介したメカニズムとしては、血管内皮細胞で血管増殖因子の分泌、内皮細胞の管腔形成が促進し、核内受容体、NF-κBやAP-1が活性化される。また血管内皮細胞にAGEsを添加するとTNF-αのm-RNAが誘導されるとの報告があり、AGEsの局在が内皮細胞に対する血管新生誘導の一因になっている。
以上のようにAGEsは神経障害の発症進展に深く関与しており、直接神経機能を障害するだけでなく、神経障害の発症因子にも大きな影響を与えている。このためAGEsの生成抑制は神経障害の治療上重要な課題である。
糖尿病性網膜症
糖尿病の眼合併症には、基底膜異常から遷延性角膜びらんを繰り返す角膜症、若年期にも発症する白内障、網膜症がある。特に網膜症は糖尿病患者の約20%に認められ、年間3,000~4,000人が網膜症による社会的失明に陥っている。網膜症の発症機序はジアシルグリセロール(DAG)-プロテインキナーゼC(PKC)経路の活性化、ポリオール代謝経路の活性化、酸化ストレス、糖化反応の亢進によるAGEsの蓄積が知られている。
高血糖は血漿タンパク質のみならず基底膜などの細胞外マトリックスを糖化しAGEsを生成させる。網膜症の発症・進展と血清・皮膚・角膜のAGEs濃度が正相関するという報告が多数あり、AGEsは網膜血管細胞に対する直接作用、受容体を介する作用、サイトカインの上昇に影響すると考えられている。AGEsの生成過程における活性酸素の産生・蓄積およびスーパーオキシドジスムターゼ(SOD)の糖化による酸化ストレス消去能の低下は、網膜血管内皮細胞表面でのヘパラン硫酸プロテオグリカンの減少による機能障害や、周皮細胞の酸化ストレスによる損傷に伴う石灰化促進を伴う細胞死を起こさせる。細胞外で生成したAGEsはRAGEの発現を促進しPKCを活性化させる。さらにAGEsはサイトカインの一つである血管内皮増殖因子(VEGF:vascular endothelial growth factor)の発現を亢進し、網膜血管の透過性亢進、微小血管の閉塞、血管新生を誘導し、網膜症の病態形成に関与している(図3)。
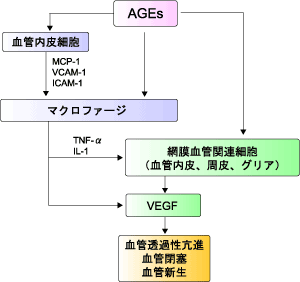
図3. AGEsによる網膜症発症のメカニズム
VEGF: 血管内皮増殖因子
網膜症は光凝固や硝子体手術の進歩によって完全失明に至る症例を減少させることができるようになったが、外科的治療による視力維持・改善にも限界がある。このためAGEsの生成抑制剤をはじめとした薬物による早期治療法の確立が待たれている。
糖尿病性腎症
糖尿病性腎症は日本における透析導入原因の第1位で、透析療法に至った糖尿病患者の5年生存率が約50%と報告されている。高血糖による血行動態の変化としては、腎症の発症早期より糸球体過剰濾過と糸球体高血圧が認められる。これらの変化は血管内皮細胞の障害を介して、メサンギウム細胞(糸球体外血管間膜)の機能異常を引き起こす。代謝因子の変化としては、DAG-PKC-MAPK(mitogen-actiated protein kinase)の活性化、タンパク質の糖化亢進によるAGEsの蓄積、酸化ストレスの亢進があり、これらが相互に影響しながら腎症発症・進展に関与していると考えられている(図4)。

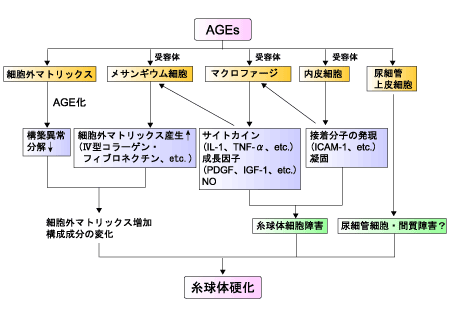
図4. AGEsによる腎障害のメカニズム
ストレプトゾトシン誘発糖尿病ラットの腎臓組織中AGEsは、コントロールと比べて45倍多く蓄積し、2型糖尿病モデル(OLETF)ラットでも糸球体でのCML局在が確認されている。さらに糖尿病性腎症患者の血中AGEs濃度増加およびクレアチニンとの正相関や、硬化糸球体、細動脈硬化病変、結節性病変でのピラリン、CML の蓄積が報告されている。
高血糖の継続は細胞外マトリックスをAGEs化してタンパク質結合能を変化させ、細胞組織の構築異常を引き起こし、糸球体透過性が亢進する。また細胞外マトリックスの主要構成成分であるⅣ型コラーゲンが糖化を受けるとプロテアーゼによる分解が遅延し、細胞外マトリックスの合成促進と相まって、その増加をきたして糸球体硬化の進展に繋がっている。さらにメサンギウム細胞、内皮細胞、上皮細胞、マクロファージにはAGE受容体(RAGE)があり、炎症性サイトカインによるNF-κBの発現が糸球体の透過性変化やメサンギウム領域の拡大に関与して糸球体細胞を障害すると推定されている。
以上のように高血糖に基づくタンパク質糖化およびAGEs蓄積は糖尿病性腎症の成因の一つであり、AGEsの生成抑制が腎症発症・進展に重要になっている。
参考文献
-
- 繁田幸雄ら(編), 蛋白の糖化 AGEの基礎と臨床, 163pp, 医学書院, 1997.
- 山岸昌一(編), AGEs研究の最前線 糖化蛋白関連疾患研究の現状, 231pp, メディカルレビュー社, 2004.
- 後藤由夫(監), 糖尿病セミナー 糖尿病とは「基礎編」, 10pp, 創新社, 2001.
- Kusunoki et al, Relation between serum 3-deoxyglucosone and development of diabetic microangiopathy. Diabetes Care, 26(6), 1889-1892, 2003.
糖化と抗糖化 詳細目次
- 生体内糖化反応とAGEs
- 生体中糖化反応のモニタリング
- AGEsとポリオール代謝経路
- 糖化と糖尿病合併症
- 糖化と動脈硬化
- 糖化と骨疾患、アルツハイマー病
- 糖化と皮膚老化
- 糖化反応阻害剤(合成化合物、既存医薬品)
- 糖化反応阻害剤(ビタミン類、その他の物質)
- 天然物中の糖化反応阻害成分
- アンチエイジングと抗糖化
– 学術術情報一覧