学術情報
糖化ストレスとアルツハイマー病
糖化ストレスとアルツハイマー病
認知症とアルツハイマー病
日本の認知症高齢者数は2012年において462 万人と推計されている 1) 。その後も増加し、2025年には675 万人(有病率18.5%)、2050年に797 万人(有病率21.1%)に達すると推定されている。2025年には65 歳以上の高齢者の約5人に1人が認知症有病者になる計算になり、誰もが関わる可能性の高い疾患である。(各年齢層の認知症有病率が2012 年以降一定と仮定した場合)
認知症は一度正常に達した認知機能が後天的な脳の障害によって持続性が低下し、日常生活や社会生活に支障をきたすようになった状態を示し、それが意識障害のないときにみられる症状をいう 2) 。世界保健機関(WHO)による国際疾病分類第10版(ICD-10)では「通常、慢性あるいは進行性脳疾患によって生じ、記憶、思考、見当識、理解、計算、学習、言語、判断等多数の高次脳機能の障害からなる症候群」とされる 2) 。認知症はアルツハイマー型認知症(アルツハイマー病)(Alzheimer’s disease:AD)、脳血管性認知症(vascular dementia:VD)、レビー小体型認知症(dementia with Lewy bodies:DLB)、他疾患による認知症の4種類に分類される。
島根県海士町での調査では65歳以上の方943名のうち104名に認知症が認められ、AD 63.5%、VD 15.4%、DLB 4.8%、パーキンソン病を伴う認知症(Parkinson’s disease dementia:PDD)6.7%、進行性核上性麻痺(progressive supranuclear palsy:PSP)1.9%、前頭側頭型認知症(frontotemporal lobar degeneration:FTLD)0.96%で、ADが直接の原因となって起こる認知症が60%以上を占めていた 3) 。
ADの症状には「軽度認知機能障害」(mild cognitive impairment:MCI)と「行動・心理症状」(周辺症状)(behavioral and psychological symptoms of dementia:BPSD)がある。MCI は記銘力障害、失見当識、判断力低下、失語、失行、失認などで、これらをまとめて中核症状(core symptom)という。BPSDは興奮、叫声、不穏、焦燥、徘徊、社会文化的に不適切な行動、性的脱抑制、収集癖、暴言、つきまとい、不安、抑うつ、妄想、幻覚などが含まれる。さらにBPSDは、興奮、易刺激性、焦燥、幻覚、妄想などの陽性症状(positive symptoms)と、無気力、無関心、抑うつなどの陰性症状(negative symptoms)に分けられる。また認知症患者の約60~90% は複数のBPSD 症状を有し、特に無関心、興奮、易刺激性、抑うつなどを有する頻度が高い 4) 。
ADの神経病理学的な特徴は、脳内の海馬(hippocampus)や大脳皮質(cerebral cortex)に萎縮(atrophy)が見られ、神経細胞の脱落、老人斑(senile plaques)や神経原線維変化(neurofibrillary tangle:NFT)の沈着が顕微鏡的に広範に認められる(図1) 5) 。NFTは電子顕微鏡像として螺旋状線維凝集物(paired helical filament:PHF)と呼ばれる特異な繊維束である。老人斑やPHFの主要構成成分には、アミロイドβ蛋白(amyloid-β protein:Aβ)と高度にリン酸化したタウ蛋白(tau protein)が同定されている。タウ蛋白は神経軸索内に存在する分子量約5万の微小管(microtubule)結合蛋白であり、微小管の重合を促進し、安定化する機能を有している。微小管は細胞骨格を形成し細胞内の蛋白や細胞内小器官で輸送のレールとして機能している。この細胞内輸送機構はタウ蛋白がリン酸化されると微小管が不安定になって抑制される。
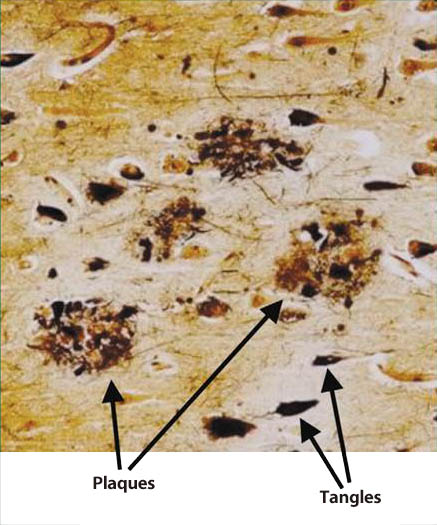
図1. アルツハイマー病患者脳の老人斑と神経原線維変化 (5)
plaques:老人斑, tangles:神経原線維変化
またAβの沈着である老人斑はNFTと比べてAD に対する疾患特異性が高いこと。主に非線維性Aβの沈着である、びまん性老人斑(diffuse plaques)はAD 脳の最初期病変であること。常染色体性優性遺伝形式をとる家族性AD のなかには、Aβ前駆体(amyloid precursor protein:APP)の点突然変異(point mutation)や重複(duplication)が疾患と連鎖して見出されていること。Aβのうち特に重合したAβ凝集体(amyloid-β aggregate)が神経毒性を有すること。これらの特徴からAβの脳内沈着はADの発症機序においてタウ蛋白よりも前段階であり、ADの病因と密接に関連した現象と推測されている。Aβを中心に据えたAD 発症機構説は「アミロイドカスケード仮説(amyloid cascade hypothesis)」と呼ばれる 6) 。
またAD患者は正常対照群と比較して大脳皮質においてコリンアセチルトランスフェラーゼ(choline acetyltransferase:ChAT)活性が低下していること。AD病患者の大脳皮質ChAT 活性と認知機能スコア(global cognitive score)は相関していること。ADでは前脳基底部(basal forebrain)のマイネルト基底核(nucleus basalis of meynert:NBM)にアセチルコリン作動性神経細胞の顕著な脱落が見られ、前脳基底部に存在する中枢神経系細胞群から大脳皮質や海馬に投射(projection)するアセチルコリン作動性神経経路には障害があること。AChE 阻害剤やニコチン(nicotine)は脳内アセチルコリン系神経伝達を促進すること。一方、アトロピン(atropine)やスコポラミン(scopolamine)の投与によって脳内のアセチルコリン系神経伝達を遮断すると、学習、記憶行動に影響を及ぼすことが示されている。これらのことからアセチルコリン作動性神経の障害は、学習記憶を含む認知機能に深く関係していることが裏付けられている。これらアセチルコリン産生不全によるADの発症機構説は「コリン作動性仮説(cholinergic hypothesis)」と呼ばれる 7) 。
さらにAD患者の老人斑沈着は神経細胞の脱落と相関していないという観察結果から、認知症の主要原因物質がタウ蛋白であり、微小管安定化因子であるタウ蛋白が消失し、神経細胞の軸索に螺旋状線維凝集物(paired helical filament:PHF)を形成してNFTとなり、細胞骨格の変性につながることが病因とする説もある。このAD発症機構説は「タウ仮説(tau hypothesis)」と呼ばれる 8-9) 。しかし、タウ蛋白の過剰リン酸化とPHFの形成のどちらが先に起こるのかは議論がある。
糖化は蛋白変性、架橋結合の亢進およびRAGEを介した炎症誘導の観点から、主にアミロイドカスケード仮説とタウ仮説に関連する。
アルツハイマー病と糖尿病
糖尿病の代表的な合併症には神経障害(neuropathy)、網膜症(retinopathy)、腎症(nephropathy)、心血管病変(cardiovascular disease)などがある。さらに糖尿病患者では認知症発症率が高いことも知られている(図2) 10) 。ロッテルダム大学による大規模疫学研究(Rotterdam study)では、平均年齢55歳以上の6370名(2型糖尿病患者(2DM)692名、非糖尿病患者5678名)について認知症との関係が1990年から約2年間調査された 11) 。その結果、認知症は126名(2.0%)が発症し、そのうち27.0%が2DMであった。認知症発症者126名のうち89名(70.6%)はADであり、認知症を発症しなかった6244名のうち10.5%が2DMであった。この結果、2DMは非糖尿病患者と比べて、ADリスクで1.9倍高いことが推定された。

図 2. 糖尿病患者と健常者の年齢別有病率 (Kimura R, et al 10) )
◆:糖尿病患者, ■:健常者, * ; P < 0.01, ** ; P < 0.001
糖代謝能と認知症との関係については福岡県久山町で長期間にわたる調査が行われている 12) 。認知症のない60歳以上の高齢者1017名(女性580名、男性487名)を1988年から2003年まで15年間追跡調査し、232名(女性153名、男性79名)について開始時の75 g経口ブドウ糖負荷試験(oral glucose tolerance test:OGTT)による糖代謝能評価結果と認知症発症との関係が調査された。その結果、認知症リスク(性・年齢調整後)は、耐糖能正常群(control)を1.0(n=115)としたとき、空腹時血糖異常群(impaired fasting glycemia)0.63(n=13)、耐糖能異常群(impaired glucose tolerance)1.35(n=63)、糖尿病型群(diabetes)1.74(n=41)であり、糖代謝能が低下しているほど高リスクであった。糖尿病および糖代謝能が認知症発症に関与していることが推定されている。
アルツハイマー病とAGEs
AD患者脳の神経病理学的な特徴は脳萎縮、老人斑、神経原線維変化である。老人斑はアミロイドβ蛋白(amyloid-β protein:Aβ)で構成され、約40個のアミノ酸で構成される分子量4300~4500のペプチド(Aβ40, Aβ42)である。Aβはβ-及びγ-セクレターゼ(secretase)の働きによりアミロイド前駆体蛋白(amyloid-β protein precursor:APP)から切り出される。ADではAβが凝集して不溶性の線維を形成してアミロイドとして沈着する。Aβはグルコース存在下でインキュベートすることで糖化しAGE化する(AGE-Aβ)。Aβのアミノ酸配列中には16と28番目にリジン残基(Lys:K)、5番目にアルギニン残基(Arg:R)があり、これらのアミノ酸配列部位が糖化に関与する可能性がある。またAβはグルコース存在下でインキュベートすると凝集する。Aβ-グルコース溶液中にAGE-Aβを添加すると、AGE-Aβを添加しない場合と比べて凝集が強く促進された 13) 。これらのことからAGE-AβはAβの凝集を促進する「シード(seed)」として作用することが推定された。また前頭葉の老人斑を含む組織の蛋白分画中AGEs量は、健康な高齢者と比べてAD患者で3倍多かった 14) 。一方、Aβ-グルコース反応系に糖化反応阻害剤であるアミノグアニジン(aminoganidine)を添加してインキュベートするとAβの凝集形成が抑制された 15) 。また老人斑中にはペントシジン、ピラリン、CMLの存在が確認されている 14-15) 。これらのことから、糖化によるAβのAGE化は蛋白架橋の形成、Aβの凝集を促進する因子のひとつとして関係することが推定される。
AD患者の脳から得られた螺旋状線維凝集物(paired helical filament:PHF)中のタウ蛋白はAGE化している。しかし非AD患者または認知症のない脳から得られた可溶性のタウ蛋白はAGE化がみられない。タウ蛋白のin vitro実験系において、糖化したタウ蛋白はPHF様の線維を形成する。しかし糖化していないタウ蛋白は線維形成しない 16) 。PHF中にはペントシジン、ピラリン、CMLが含まれる。またタウ蛋白のアミノ酸配列中には13個のリジン残基があり、このうち6個が糖化している 17) 。タウ蛋白の糖化部位は微小管との結合部位にあたるため、結合機能が障害される。糖化したタウ蛋白は活性酸素、IL-6などの酸化ストレスを誘導し、神経細胞機能に障害を受ける 18) 。糖化したタウ蛋白はアミロイド前駆体蛋白(amyloid-β protein precursor:APP)とAβの生成を誘導する。タウ蛋白はリン酸化や糖化によって微小管結合機能が低下し、タウ蛋白本来の機能である細胞内輸送能が低下する。このため細胞はAPPを細胞外に分泌できなくなり、細胞内にAPPが蓄積する。これらのメカニズムによりAβやタウ蛋白の糖化はAβの沈着・凝集に促進的に作用すると同時に、タウ蛋白の沈着を亢進する。
さらにAD患者の脳内に存在するミクログリア(microglia)、アストロサイト(astrocyte)にはペントシジン、CMLおよびAGE受容体であるRAGE(receptor for AGEs)が存在し 19) 、健常な高齢者と比べて有意に増加しているミクログリアは中枢神経系に存在する常在性マクロファージ(macrophage)とも呼ばれる細胞の一種で、ニューロンの除去や修復機能などの機能を有する 20) 。一方、ミクログリアの活性化により腫瘍壊死因子(TNF-α)、インターロイキン(IL)-1β、インターフェロン(IFN)などの炎症性サイトカインの産生が認められ、神経細胞障害性に働く可能性が示唆されている 21) 。アストロサイトは脳と脊髄に特異的に存在する星型のグリア細胞で、中枢神経系の機能維持において血流調節、神経細胞へのエネルギー供給、シナプス機能への関与、神経伝達物質の調整などを行っている 22) 。ミクログリア培養細胞にAGE化鶏卵アルブミン(chicken egg albumin-AGE)を添加するとRAGEを介したNO(nitric oxide)、IL-6、TNF-αなどの産生がみられ、炎症が惹起された 23) 。またラット大脳皮質神経細胞(cortical neuronal cell)培養系に各種AGEsを添加した実験では、グリセルアルデヒド(glyceraldehyde)由来AGEs(AGE-2)が、アマドリ化合物(amadori products)(AGE-1)、グリコールアルデヒド(glycolaldehyde)由来AGEs(AGE-3)、メチルグリオキサール(methylglyoxal)由来AGEs(AGE-4)、 グリオキサール(glyoxal)由来AGEs(AGE-5)と比べて強いアポトーシス(apotosis)誘導が確認された 24) 。このときCML、CEL は細胞死を誘導しなかった。これらのことから脳内においてAβや老人斑の蓄積に伴うAGEsの生成蓄積は、Aβの神経毒性獲得、RAGEを介した炎症およびアポトーシスを誘導し、ADの病態である神経細胞死の促進、増強への関与が推定された(図3) 25) 。
これらのことから糖化によるAGEsの生成や蓄積を低減する物質の活用は、ADの発症を防ぐ対策となる可能性が示唆されている 26-27) 。
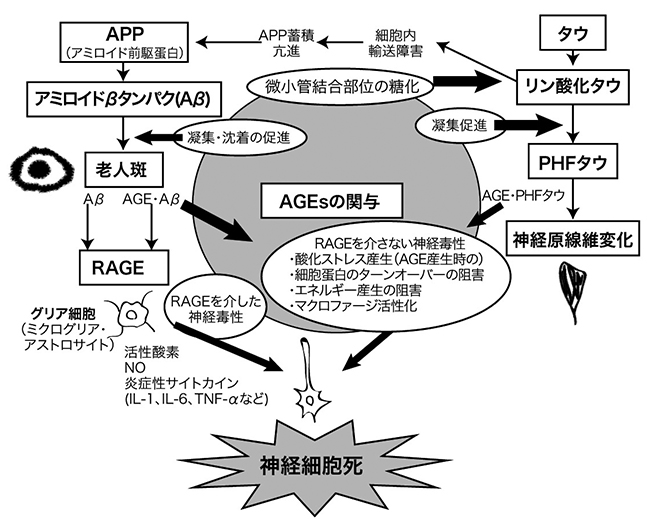
図3. AGEsのアルツハイマー病への関与 (佐々木信幸 25) )
参考文献
-
- 二宮利治:日本における認知症の高齢者人口の将来推計に関する研究総括研究報告書, 厚生労働省; 2015.
- 認知症疾患治療ガイドライン2010 ; 日本神経学会監修.
- Wada-Isoe K, et al.:Neuroepidemiology. 2009; 32: 101–106.
- Ileda M et al.:J Neurol Neurosurg Psychiatry. 2004; 75: 146–148.
- Blennow K, et al.:Lancet. 2006; 368: 387–403.
- Karran E, et al.:Nature Reviews Drug Discovery. 2011; 10: 698-712.
- Francis PT, et al.:J Neurol Neurosurg Psychiatry. 1999; 66: 137–147.
- Mudher A, et al.:Trends Neurosci. 2002 ; 25(1): 22-26.
- Maccioni RB, et al.:Archives of Medical Research. 41; 2010: 226-231.
- Kimura R, et al.:Psychogeriatrics. 2008; 8: 73-78.
- Ott A, et al.:Neurology 53(9); 1999: 1937-1942.
- Ohara T, et al.:Neurology. 2011; 77: 1126–1134.
- Vitek MP, et al.:Proc Natl Acad Sci USA. 1994; 91: 1994.
- Smith MA, et al.:Proc Natl Acad Sci USA. 1994; 91: 5710-5714.
- Horie K, et al.:Biochem Biophys Res Commun, 1997; 236: 327–332. 1997
- Ledesma MD, et al.:J Biol Chem. 1994; 269: 21614-21619.
- Nacharaju P, et al.:J Neurochem. 1997; 69: 1709-1719.
- Yan SD, et al.:Proc. Nati. Acad. Sci. USA: 1994: 7787-7791
- Srikanth V, et al.:Neurobiology of Aging. 2011; 32: 763–777.
- Takeda A, et al.:Acta Neuropathol. 1998; 95: 555–558.
- 錫村明生:福岡医誌. 100; 243-247 2009
- Hamby ME, et al.:Neurotherapeutics. 2010; 7: 494-506.
- Dukic-Stefanovic S, et al.:Journal of Neurochemistry. 2003; 87: 44–55.
- Takeuchi M, et al.:J. Neuropathol. Exp. Neurol. 2000; 59: 1094–1105.
- 佐々木信幸:AGEsとアルツハイマー病, AGEsの研究の最前線. 2004: 171-177.
- Dukic-Stefanovic C, et al.:Biogerontology. 2001; 2: 19–34.
- Munch G, et al.:Biochem Soc Trans. 2003; Pt 6: 1397-1399.